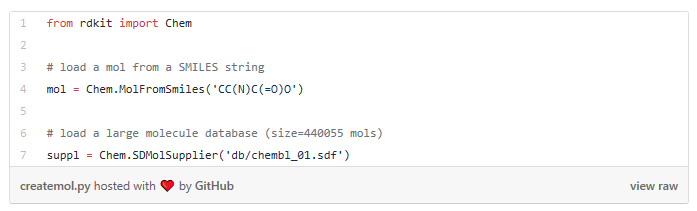
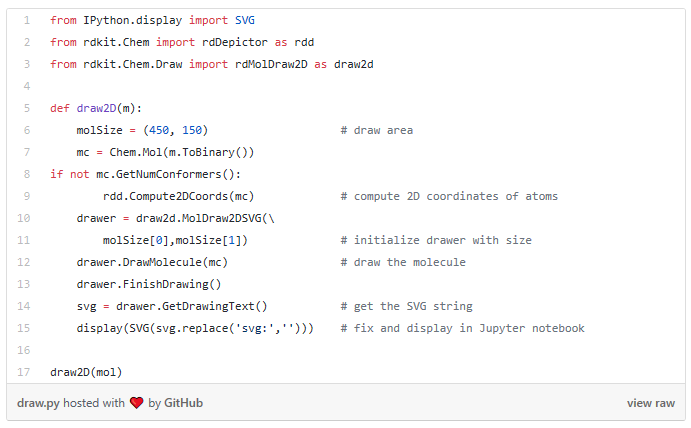
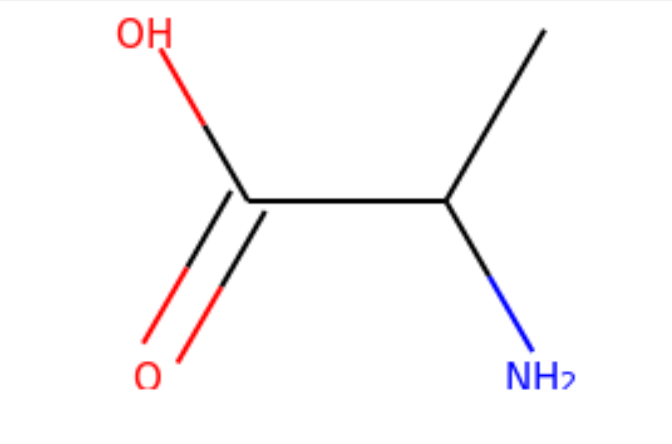
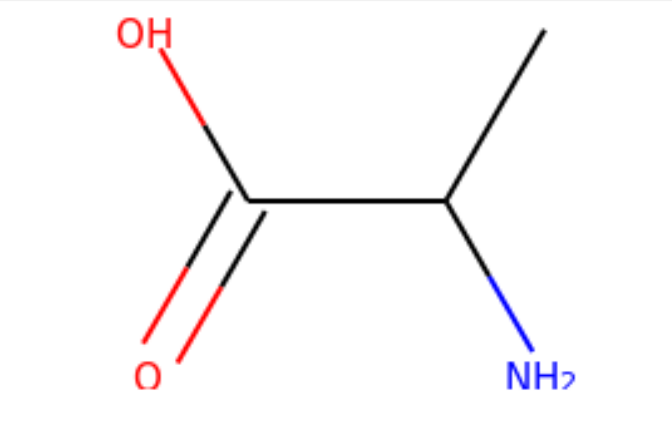
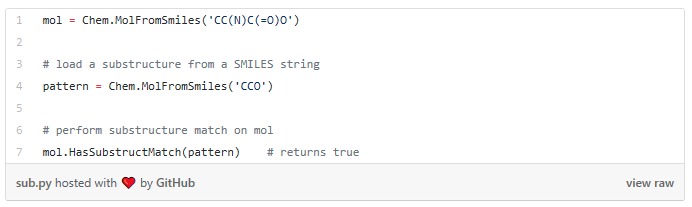
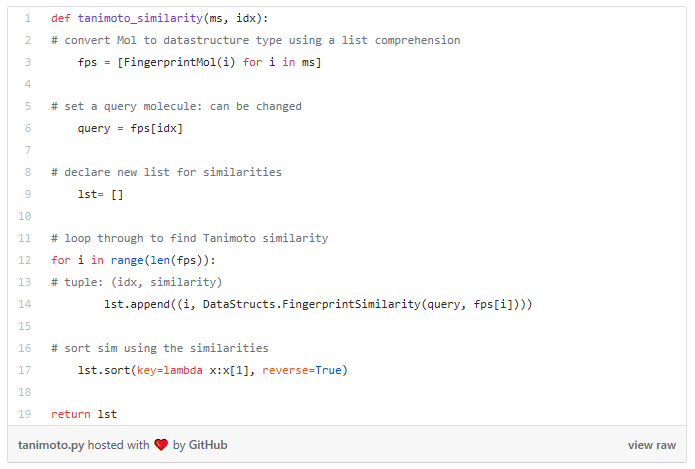
I started my very first internship at GSI Technology and my first project was on cheminformatics — it was on performing similarity search on molecules. Similarity search can be done in various ways, and RDKit is one popular toolkit that helps you calculate similarities of molecules directly. Additionally, my mentor also mentioned exploring more ways to do similarity search on molecules, such as graph embeddings, but we will save that for a later post. Drug Discovery is trending in Data Science! When I first scrolled through the RDKit “Getting Started” package, I was initially confused by all the numbers and chemical formulas. But I’m not a chemist! And all the functions looked so complicated, for example, MolFromSmiles(), MolToMolBlock(), and so many more. However, as I went through it step by step, it was not as complex as I thought it was. It was just that the chemical terms were intimidating. Now, I’m going to show you how simple it is to use RDKit. First, let’s look at a couple of ways to create molecules using RDKit: The first example creates a molecule from what’s called a SMILES string. The letter in the string stands for an atom — C is carbon, O is oxygen, and N is nitrogen. The other symbols in the string describe the bonds between the atoms. The second example creates a list of molecules from a molecule database. The database I used is openly available from the chEMBL site. Now that we know how to load molecules, we might want to visualize them. In RDKit, that’s easy. There are lots of different ways and examples on the Internet about displaying molecules, I’m using one of them (RDKit in Jupyter,) and this is the code I copied from it. And this is what the molecule loaded from the SMILES string looks like: mol = CC(N)C(=O)O Substructures are very important in cheminformatics. A substructure is just a part of a molecule. For example, look at the molecule below to the left. I’ve highlighted just one substructure of the molecule in yellow, which is isolated and shown to the right. Why are substructures important? left: CC(N)C(=O)O; right:CCO It turns out that certain substructures determine how a molecule behaves as a drug. And so molecules that share the same key substructures are likely to behave in a similar way. RDKit helps us match substructures between molecules. Notice in the example above, since mol contains the substructure pattern, the function HasSubstructMatch() returns true. Now that we can load and display molecules, let’s move on to similarity search. Why is similar search important for molecules? Here is one use case. A pharmaceutical company is designing a new drug for pain relief. Because synthesis of new drugs is very expensive, they want to start the process with the best “seed” molecules — the ones with the highest chance of success. One technique would be to take a molecule that has known pain relief properties and perform a similarity search against a large “virtual” molecule database. There is a good chance that the top matches also have the desired properties. In this way, the company starts the expensive and time consuming drug discovery process with the best leads. RDKit provides tools for different kinds of similarity search, including Tanimoto, Dice, Cosine, Sokal, Russel… and more. Tanimoto is a very widely use similarity search metric because it incorporates substructure matching. Here is an example: Using that function I performed a query using the molecule shown on the top. On the bottom are the top 6 matches. top: the query; bottom: top 6 matches including the query Notice that all the matches look similar to the query. The first match is the query itself. This makes intuitive sense since a molecule should be most similar to itself (but in many similarity searches the query won’t already exist in the database.)Introduction

What is RDKit?
Visualizing Molecules
Substructures
Similarity Search
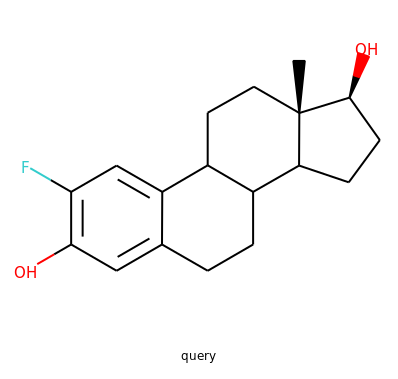
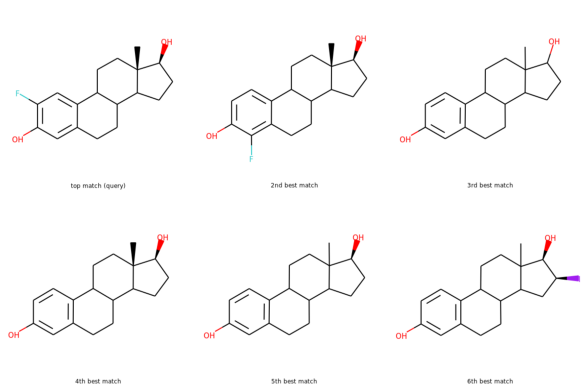
When I heard about my first assignment, I thought it would take me forever to understand and complete it, but it turned out RDKit wasn’t as hard as I imagined it would be!
In my next blog, I will be showing a different similarity search on molecules using other techniques and I will compare them to Tanimoto similarity search.